ISSN: 1449-2288International Journal of Biological Sciences
- Current issue
- Volume 20; 2024
- Volume 19; 2023
- Volume 18; 2022
- Volume 17; 2021
- Volume 16; 2020
- Archive
- Advance articles
- Cover images
- Index & coverage
- Cover suggestion
- Special issues
Introduction
The Hypothesis
The Hypothesized Main Steps
The Main Differences Between Our...
The Significance of Our...
Acknowledgements
References

Int J Biol Sci 2008; 4(4):246-258. doi:10.7150/ijbs.4.246 This issue Cite
Research Paper
Bad seeds produce bad crops: a single stage-process of prostate tumor invasion
Yan-gao Man1,2 ![]() , William A. Gardner1
, William A. Gardner1
1. American Registry of Pathology and Armed Forces Institute of Pathology, Washington DC, USA
2. Jilin University, Changchun, Jilin, China
Abstract
It is a commonly held belief that prostate carcinogenesis is a multi-stage process and that tumor invasion is triggered by the overproduction of proteolytic enzymes. This belief is consistent with data from cell cultures and animal models, whereas is hard to interpret several critical facts, including the presence of cancer in “healthy” young men and cancer DNA phenotype in morphologically normal prostate tissues. These facts argue that alternative pathways may exist for prostate tumor invasion in some cases. Since degradation of the basal cell layer is the most distinct sign of invasion, our recent studies have attempted to identify pre-invasive lesions with focal basal cell layer alterations. Our studies revealed that about 30% of prostate cancer patients harbored normal appearing duct or acinar clusters with a high frequency of focal basal cell layer disruptions. These focally disrupted basal cell layers had significantly reduced cell proliferation and tumor suppressor expression, whereas significantly elevated degeneration, apoptosis, and infiltration of immunoreactive cells. In sharp contrast, associated epithelial cell had significantly elevated proliferation, expression of malignancy-signature markers, and physical continuity with invasive lesions. Based on these and other findings, we have proposed that these normal appearing duct or acinar clusters are derived from monoclonal proliferation of genetically damaged stem cells and could progress directly to invasion through two pathways: 1) clonal in situ transformation (CIST) and 2) multi-potential progenitor mediated “budding” (MPMB). These pathways may contribute to early onset of prostate cancer at young ages, and to clinically more aggressive prostate tumors.
Keywords: prostate carcinogenesis, tumor invasion, pre-invasive lesions
Introduction
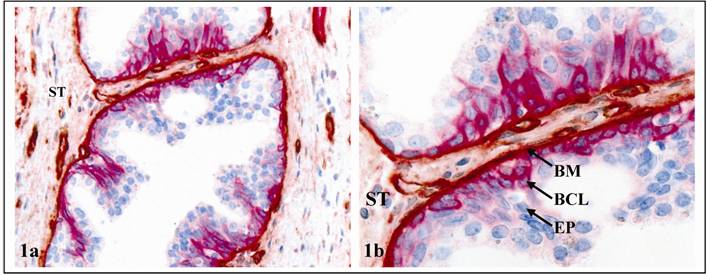
The normal prostate luminal cells, which are the histological origin of most prostate malignancies, are physically separated from the stroma by the basal cells and basement membrane (BM). Basal cells are joined by intercellular junctions and adhesion molecules, constituting a continuous sheet encircling luminal cells [1-2]. The BM is composed of type IV collagen, laminins, and other molecules, forming a continuous lining surrounding and attaching to the basal cell layer [3-4]. The epithelium is normally devoid of blood vessels and lymphatic ducts, and totally relies on the stroma for its metabolic and even survival needs. Because of these structural relationships (Fig 1), the disruption of both the basal cell layer and the BM is a pre-requisite for prostate tumor invasion or metastasis.
It is a commonly held belief that human prostate carcinogenesis is a multistage process, progressing sequentially from normal to hyperplasia, to prostatic intraepithelial neoplasia (PIN), and to invasive or metastatic stages [5-8]. Progression from PIN to invasion is believed to be triggered by overproduction of proteolytic enzymes primarily by cancer cells, which cause degradation of the BM [9-10]. These theories are consistent with results of studies in tissue cultures and animal models, whereas are hard to interpret the following critical facts:
- Our previous studies revealed that some healthy men between 19 and 29 years old had a spectrum of proliferative lesions, including hyperplasia, PIN, and incipient adenocarcinoma [11-13].
- Recent studies detected a DNA phenotype that is identical to that of invasive prostate cancer in some “healthy” men, and in morphologically normal prostate tissues adjacent to prostate cancer [14-17].
- A vast majority of PIN express high levels of proteolytic enzymes, while only 10-30% of untreated PIN progress to invasive lesions during patients' lifetime [18-21]. Unfortunately, none of the current approaches could predict which PIN lesions will progress [22-25]. The only established approach to monitor PIN progression is repeat biopsy [22-25], which is costly and painful.
Together, these facts argue that alternative pathways may exist, because if the linear model of tumor progression and invasion is universally applicable, the normal prostate tissues of healthy subjects should not harbor malignant structural abnormalities. Since over 90% of prostate cancer related mortality result from invasion-related illness, and the incidence of PIN could be up to 16.5%-25% in prostate biopsies [24-28], there is an urgent need to uncover the intrinsic mechanism of tumor invasion.
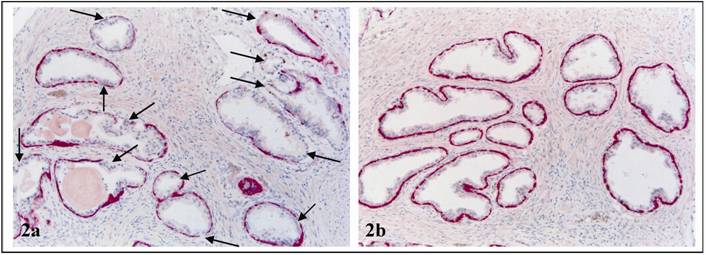
Promoted by the fact that the basal cell layer is the sole source of tumor suppressor p63 and maspin [29-32], and that degradation of basal cell layers is a pre-requisite for tumor invasion, our resent studies have attempted to identify early signs of basal cell degradation. Our initial study examined the physical integrity of basal cell layers in 50 patients with co-existing pre-invasive and invasive prostate tumors. Of 2,047 ducts and acini examined, 197 were found to harbor focal disruptions (the absence of basal cells resulting in a gap greater than the combined size of at least 3 basal cells) in their basal cell layers. The frequency of focal basal cell layer disruptions (FBCLD) varied from none in 22 (44%) cases to over 1/3 of the ducts or acini with FBCLD in 17 (34%) cases (Table 1) [33].
Frequencies of focal basal cell layer disruptions among cases
| Case number | No disruptions | < 30% disruptions | ≥30% disruptions | p |
|---|---|---|---|---|
| 50 | 22 (44%) | 11 (22%) | 17 (34%) | < 0.01 |
Structural relationships among the epithelium (EP), basal cell layer (BCL), basement membrane (BM), and stroma (ST). Section was double immunostained for the BM (brown) and BCL (red). a: 100X b: a higher (300X) magnification a.
Different frequencies of FBCLD among cases. Double immunostained for CK 34ßE12 (red) and Ki-67 (brown). In case 1 (a), all 12 acini show FBCLD (arrows), while in case 2 (b), none of the 12 acini shows FBCLD. 300X.
Of the 17 cases with a high frequency of FBCLD, the basal cell layer had several unique alterations that were not or rarely seen in their morphologically similar counterparts in other cases [33-44]:
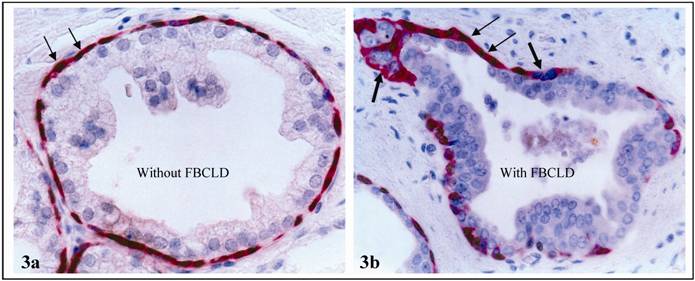
a. significantly reduced expression of tumor suppressor p63: In sections double immunostained for p63 and CK34ßE12, an average of 87% of the basal cells in non-disrupted layers expressed both molecules, while only 59% of the basal cells in focally disrupted layers had p63 expression (Fig 3; Table 2).
p63 expression in basal cell layers with and without focal disruption
| Basal cell layer status | Number of ducts or acini | Percentage of p63 (+) cells | P |
|---|---|---|---|
| With disruption | 197 | 59% | < 0.01 |
| W/o disruption | 197 | 87% |
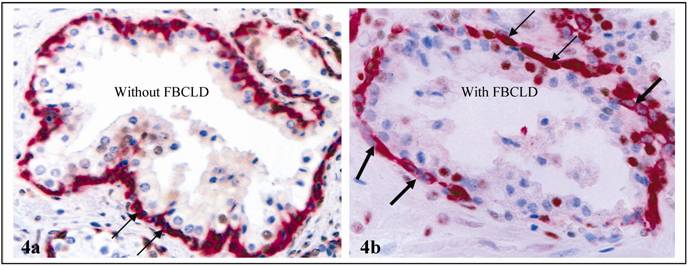
b. significantly reduced expression of proliferating cell nuclear antigen (PCNA): In sections double immunostained for PCNA and CK4ßE12, an average of 74% of the normal basal cells showed PCNA expression, but only 51% of basal cells in disrupted basal layers were PCNA positive (Fig. 4; Table 3).
PCNA expression in basal cell layers with and without focal disruption
| Basal cell layer status | Number of ducts or acini | % of PCNA (+) cells | P |
|---|---|---|---|
| With disruption | 50 | 51% | < 0.01 |
| W/o disruption | 50 | 74% |
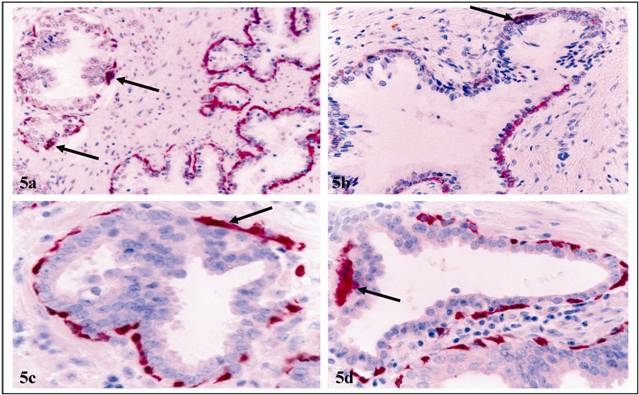
c. significantly elevated apoptosis and degeneration: Of 78 ducts and acini with FBCLD examined, 59 (75.6%) harbored apoptotic basal cells, compared to 9 (11.5%) in 78 similar structures with intact basal layers. Under high magnification, basal cells near FBCLD often had cytological signs of degeneration, including nuclear swelling, shrinkage, fragmentation, or rod-like structures of fused basal cells (Fig. 5).
Loss or reduction of p63 expression in focally disrupted basal cell layers. Sections were double immunostained for CK 34ßE12 (red) and p63 (brown). Thin and thick arrows identify cells with and without p63 expression, respectively. 400X.
Significantly reduced PCNA expression in basal cell layers with FBCLD. Double immunostained for CK34 ßE12 (red) and PCNA (brown). Thin and thick arrows identify basal cells with and without PCNA expression, respectively. 400X
Elevated apoptosis and degeneration in focally disrupted basal cell layers. Section was assessed for apoptosis (a-b) or CK34βE12 expression (c-d). Arrows identify apoptotic or degenerated basal cells arranged as rod-like structures. 300X.
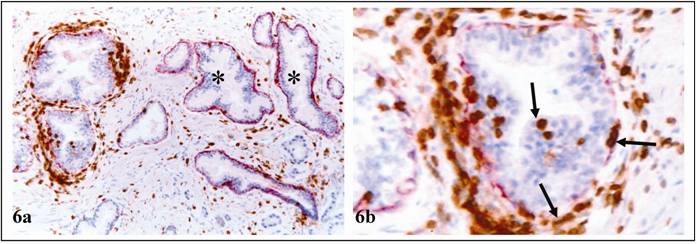
d. significantly elevated leukocyte infiltration: In sections double immunostained for CK 34ßE12 and leukocyte common antigen (LCA), most ducts and acini with FBCLD showed leukocyte infiltration, but most ducts and acini with non-disrupted layers had no leukocyte infiltration (Table 4). Most infiltrated leukocytes were located near FBCLD (Fig 6).
Leukocyte infiltration in ducts or acini with and without focal basal cell layer disruption
| Basal cell layer status | Number of ducts or acini | Number of leukocyte infiltration | P |
|---|---|---|---|
| With focal disruptions | 201 | 183 (91.0%) | < 0.01 |
| Without focal disruptions | 201 | 67 (33.3%) |
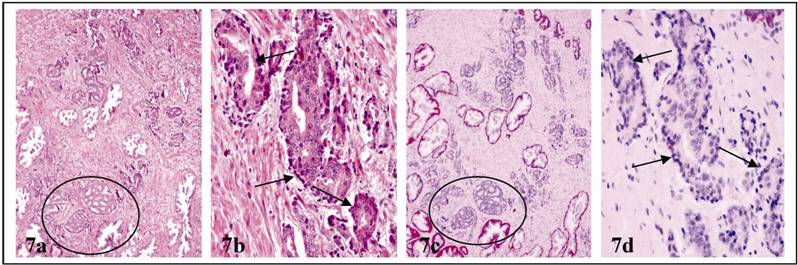
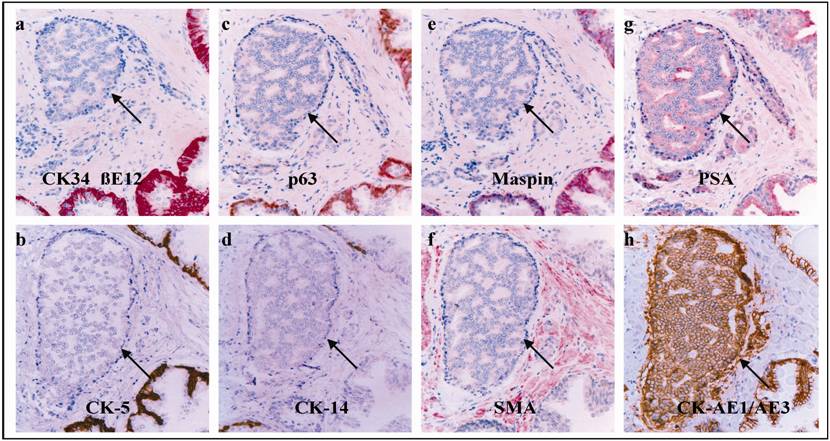
e. a total loss of the expression of all basal cell phenotypic markers: In addition to focal alterations, the entire basal cell layer in some ducts and acini showed degenerative changes. These basal cell layers were morphologically distinct, surrounding PIN or normal-appearing duct or acinar clusters (Fig 7). All the basal cells, however, lacked the expression of basal cell specific markers (Fig 8). In contrast, tumor cells in PIN and normal-appearing duct or acinar clusters had enlarged nuclei and nucleoli.
FBCLD and leukocyte infiltration. Double immunostained for CK34ßE12 (red) and LCA (brown). Arrows identify infiltrates within EP or near FBCLD. No LCA infiltration was seen in ducts with non-disrupted basal cell layers (asterisks). a; 100X. b: a higher (300X) magnification of a.
Degenerative alterations in entire basal cell layers. Two adjacent sections were H & E (a & b) or immunostained for CK34βE12 (c & d). Circles identify PIN. Arrows identify altered basal cell layers in normal appearing acinar clusters. a & c: 100X. c & d: 400X.
Morphologically distinct basal cell layers lack expression of all basal cell specific markers. Immunostained for basal (a-e), stromal (f), and epithelial (g-h) cell markers. Arrows identify altered basal cell layers. 200X.
In addition, these basal cell layers were devoid of expression of PCNA, in contrast to normal basal cells and associated tumor cells, which were strongly positive for PCNA (Fig. 9). Ducts or acini with altered basal cell layers had a significantly higher frequency of mast cell infiltration (Fig 9; Table 5).
Together, these findings suggest that the basal cell layers in these patients have significantly higher degenerative alterations. As the basal cell layer is the sole source of several tumor suppressors [29-32], degenerated basal cells are very likely to have impaired or reduced functions.
In contrast to degenerative alterations in basal cells, luminal cells overlying FBCLD showed several proliferative alterations that were not seen in their adjacent counterparts distant from the disruptions:
a. significantly elevated cell proliferation: In sections double immunostained for basal cell phenotypic and proliferation-related markers, these clusters had a significantly higher proliferation index than their counterparts without FBCLD, and most proliferating cells were located near FBCLD (Fig.10). Of 78 ducts and acini with FBCLD examined, 47 had clusters of multiple proliferating cells, compared to 8 in 78 morphologically similar ducts or acini with non-disrupted basal cell layers (Table 5).
Epithelial proliferation in ducts or acini with and without focal basal cell layer disruption
| Duct or acinar type | Total number | With proliferating cells | P |
|---|---|---|---|
| With disruption | 78 | 47 (62.5%) | < 0.01 |
| W/o disruption | 78 | 8 (10.2%) |
PNA expression and mast cell infiltration in acini with altered basal cell layers. Double immunostained for CK34 βE12 (red) and PCNA (brown) or mast cells (black). Arrows identify basal cell layers or mast cells. a & c: 100X. b & d: 400X.
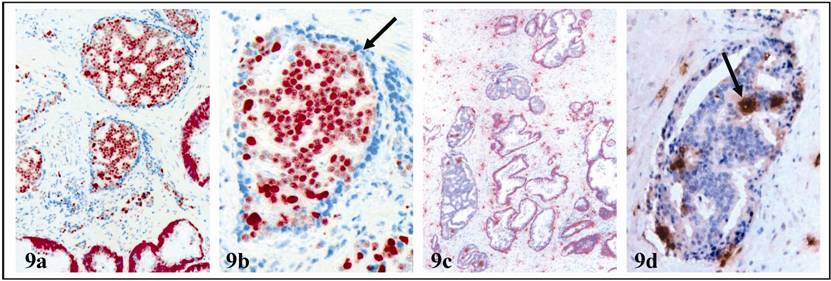
Increased proliferation in ducts with FBCLD. Sections were double immunostained for CK 34ßE12 (red) and Ki-67 (brown). Arrows identify proliferating cell clusters. Note that in a-b, KI-67 positive cells are seen in ducts with FBCLD, but not in adjacent ducts without FBCLD (square). a & c: 100X. b & d: a higher (400X) magnification of a and c, respectively.
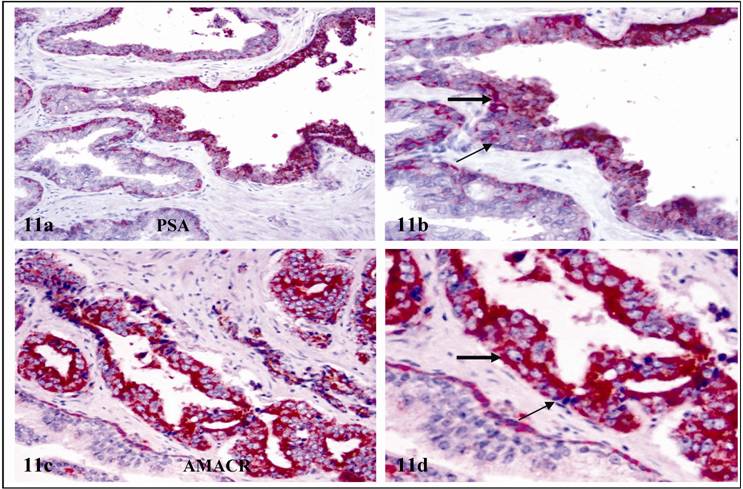
b. significantly higher expression of malignancy- and tumor invasion-related molecules: Elevated expression of prostate specific antigen (PSA) and alpha-methylacyl-CoA racemase (AMACR), are consistently seen in cells overlying FBCLD (Fig.11. a & b), and also in normal appearing ducts that lacked the expression of basal cell phenotypic markers (Fig. 11 c & d). In contrast, adjacent cells within the same duct, and adjacent ducts with intact basal cell layers were largely negative (Fig 11).
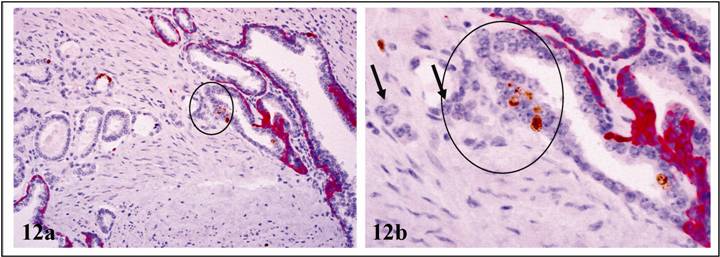
c. physical continuity with, and morphological resemblance to, invasive prostate cancer: A vast majority of these normal appearing clusters were adjacent to, or blended within, invasive cancers. In some cases, cells overlying FBCLD had significantly enlarged nuclei and nucleoli, and were in physical continuity with, or morphologically similar to, their adjacent invasive counterparts (Fig 12).
PSA and AMACR expression in cells overlying FBCLD and ducts with altered basal cells. Double immunostained for CK34 βE12 (red) and PSA (a-b) or AMACR (c-d) (brown). Thick arrows identify cells with AMACR or PSA expression. Thin arrows identify residual basal cells. a & c: 100X. b & d: a higher magnification (400X) of a & c, respectively.
Physical continuity with, and morphological resemblance to, invasive cancer. Double immunostained for CK 34ßE12 (red) and Ki-67 (brown). Circles identify proliferating cells overlying FBCLD. Note that cells overlying FBCLD are in direct continuity and similar to invasive cancer cells (arrows). a: 100X. b: a higher (400X) magnification of a.
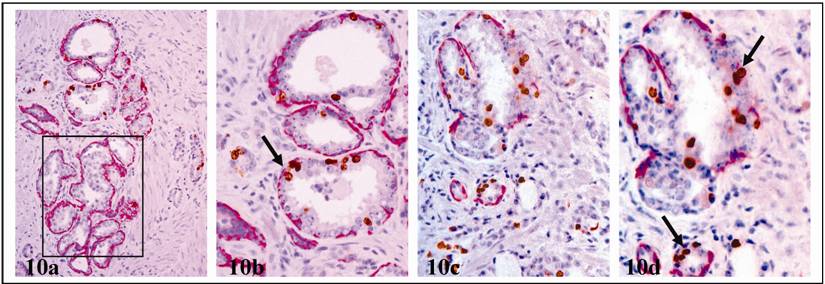
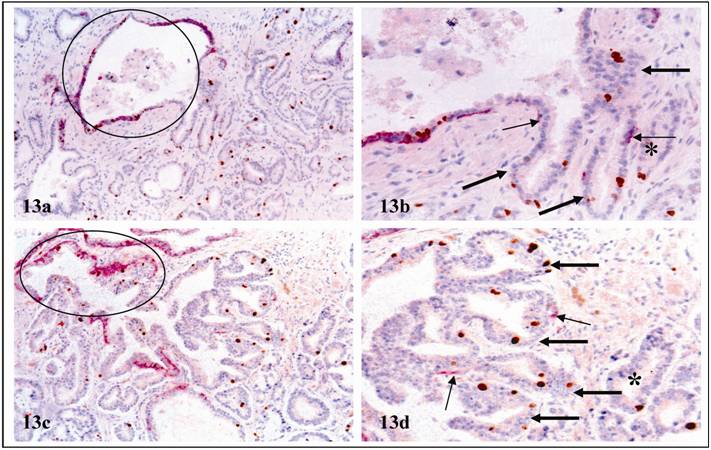
In some cases, multiple epithelial cell nests appeared to be “budding” from the same acinus or duct (Fig.13). These “budding” cells had a higher proliferation and were similar to adjacent invasive cancer cells. The only difference was that “budding” cells had residual basal cells (Fig 13, thin arrows).
d. significantly elevated expression of tumor invasion-related genes: In studies of gene expression profiling, cell clusters overlying FBCLD consistently showed significantly higher expression of cell proliferation, apoptosis, angiogenesis, immuno-response, and stem cell related genes (Fig 14; Table 6).
Cell “budding” from normal appearing ducts or acini. Double immunostained for CK34ßE12 (red) and Ki-67 (brown). Circles identify normal appearing ducts or acini. Thick arrows identify “budding” cell clusters. Thin arrows identify residual basal cells. Asterisks identify invasive cancers. a & c: 100X. b & d: a higher (400X) magnification of a and c, respectively.
Different gene expression profiles in cells overlying FBCLD and adjacent counterparts. Cells from these two locations were microdissected from frozen prostate sections, and subjected to RNA extraction, amplification, and gene expression profiling using our published protocols. Circles identify microdissected cells and differentially expressed genes.
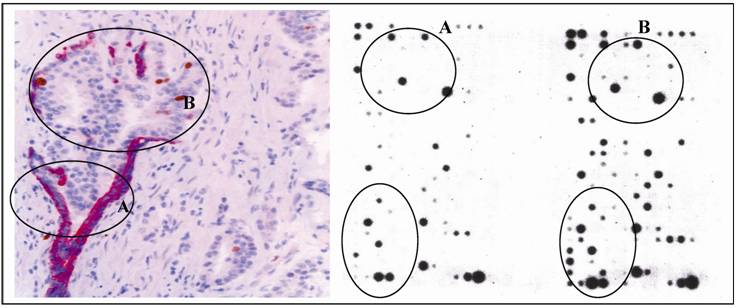
Differentially expressed genes between cells overlying FBCLD and their adjacent cells
| # | Gene name | Potential functions | Fold changes |
|---|---|---|---|
| 1 | LIF | Growth factor | 47.37 |
| 2 | MCL1 | Anti-apoptosis | 6.72 |
| 3 | TNFRSF7 | Anti-apoptosis | 7.91 |
| 4 | KIT | Stem cell lineage marker | 5.03 |
| 5 | NCOR2 | Stem cell lineage marker | 5.45 |
| 6 | ENG | Endothelial cell marker | 6.38 |
| 7 | ICAM2 | Endothelial cell marker | 12.12 |
| 8 | KRT17 | Epithelial cell marker | 7.15 |
| 9 | ITGA3 | Cell-matrix adhesion | 5.52 |
| 10 | ITGB3 | Cell-matrix adhesion | 7.14 |
| 11 | CCL2 | Chemokine, cytokine, and receptor | 14.33 |
| 12 | CX3CL1 | Chemokine, cytokine, and receptor | 6.14 |
| 13 | CCR1 | Chemokine, cytokine, and receptor | 5.19 |
| 14 | CXCR4 | Chemokine, cytokine, and receptor | 12.81 |
| 15 | TNFRSF10D | TNF receptor family | 8.20 |
| 16 | TNFRSF12A | TNF receptor family | 5.35 |
| 17 | TNFRSF25 | TNF receptor family | 8.52 |
| 18 | TIMP1 | ECM inhibitor | 5.25 |
| 19 | TIMP3 | ECM inhibitor | 7.87 |
| 20 | MMP-26 | Matrix metalloproteinase | -6.94 |
| 21 | IL10 | Interleukin and receptor | -9.50 |
| 22 | IL12RB2 | Interleukin and receptor | -7.02 |
| 23 | IL6R | Interleukin and receptor | -7.24 |
The above alterations were consistently seen in all 17 cases with a high frequency of FBCLD, while were only seen in 1 (9.1%) of the 11 cases with a low frequency of FBCLD, and in none of the 22 cases with non-disrupted basal cell layers. Together, these findings suggest that the physical and functional status of the basal cell layer significantly impact the biological presentation of associated epithelial cells. These findings also suggest that FBCLD may represent a trigger factor for prostate tumor progression and invasion.
To our best knowledge, our findings have not been previously reported by others. The most likely reasons are: [1] the enzyme theory has dominated the direction of researches in the field, and the roles of basal cells have been ignored; [2] these alterations could be seen only by double immunohistochemistry to simultaneously elucidate both basal cells and other markers. Double immunostaining, however, has not been commonly used in clinical studies.
The Hypothesis
Based on the above findings, we have proposed that these normal appearing duct or acinar clusters represent a population of maturation arrested tumor progenitors derived from monoclonal proliferation of genetically damaged primitive stem cells at early stages of the prostate morphogenesis probably by trauma, radiation, inflammation, or other factors. These clusters retain the potential for unlimited cell proliferation or multi-lineage differentiation, and could progress directly to invasive lesions through two different pathways: (1) clonal in situ transformation (CIST), and (2) multi-potential progenitor mediated “budding” (MPMB). These pathways are likely to contribute to early onset of prostate cancer at young ages, and to biologically and clinically more aggressive prostate tumors.
The Hypothesized Main Steps
Our hypothesized main steps are the followings:
- At the early stage of prostate morphogenesis, the prostate of these patients exposed to external or internal insults, such as radiation, carcinogens, localized trauma, inflammation, or other factors, which caused permanent damages in the DNA structures of some primitive stem cells.
- Localized DNA structural damages caused the inactivation of, or defects, in basal cell renewal-related genes, which impaired the basal cell replenishment process to replace the aged or injured basal cells, resulting in a “senesced” basal cell population with significantly reduced functions.
- Localized DNA structural damages also caused the inactivation of, or defects in, apoptosis-, or cell cycle control related genes in the luminal cell population, which allow these cells to escape from programmed death, to continuously proliferate, and to generate their own vascular structures.
- Deregulated proliferation in epithelial cells and impaired self-renewal in basal cells resulted in the overstretch and disassociation of the basal cell layer and the BM, which lead to focal breakdown and degeneration of these two structures.
- The degradation products of degenerated basal cells and the diffusible molecules of the overlying luminal cells function as self-epitopes to attract migration and infiltration of immunoreactive cells (IRC) or auto-antibodies into the affected sites.
- The direct physical contact between IRC and degenerated basal cells results in the discharge of the digestive enzymes from IRC, leading to the physical destruction of altered basal cell layers and the local basement membrane, resulting in a focal disruption in these structures.
- Since the EP is normally devoid of both blood vessels and lymphatic ducts, and the basal cell layer is the sole source of several tumor suppressors, a FBCLD in a given basal cell layer could lead to several focal alterations, including: a) A loss or reduction of tumor suppressors and the paracrine inhibitory functions, which allow the luminal cells to undergo elevated proliferation [45-49]. b) Alterations in the permeability for oxygen or growth factors, which selectively triggers the exit of stem or progenitor cells from quiescence, and favor proliferation of cells overlying FBCLD [50-52]. c) The exposure of luminal cells to different cytokines, which facilitates vasculogenic mimicry and tumor angiogenesis [53-54]. d) The physical contact between luminal and stromal cells augments the expression of stromal MMP, facilitating epithelial-mesenchymal transition (EMT) and cell motility [55-57]. e) The physical contact between luminal and immunoreactive cells directly cause genomic or cellular damages that trigger a cascade reaction of malignant transformation [58-63].
- These alterations could individually or collectively trigger elevated proliferation in luminal cells near FBCLD, which leads to the enlargement of FBCLD and stretching-out of the residual basal cells. Eventually, the entire basal cell layer becomes dissociated or degenerated (as those shown in Figs. 7-13), which facilitates progression of these clusters directly to invasive prostate lesions through two different mechanisms: a) Clonal in situ transformation (CIST), in which the entire luminal cell population of these clusters completely lose their surrounding structures and transform directly to a new microenvironment that favors cell motility and epithelial-mesenchymal transition. Although these cells might not possess all the properties of invasive cancer cells, the changed microenvironment may act as a second “hit” to trigger a cascade reaction of malignant transformation that rapidly alters the genetic and biochemical profiles of these cells. b) Multi-potential progenitor mediated “budding”, in which a subset of cell clusters overlying FBCLD retain the properties of primitive stem cells. Thus, they are able to constantly produce new cells and their own vascular structures through “budding” (as those shown in Fig.13), representing the source for biologically and clinically more aggressive prostate tumors.
- The above events may occur and progress immediately after the external or internal insults, leading to development of prostate cancer at young ages. On the other hand, cells of these normal appearing duct or acinar clusters may become maturation-arrested after a few cycles of cell divisions, and remain idle until a new insult [64], representing “bad seeds for bad crops” at later ages.
The Main Differences Between Our Hypothesis And Previous Theories
Based on our own and other findings, we had proposed that prostate tumor invasion is triggered by localized degeneration of the basal cells and leukocyte infiltration induced auto-immunoreactions, which selectively favor monoclonal proliferation and invasion of progenitor or stem cells overlying FBCLD [43]. Our hypothesis differs from the traditional theories of tumor invasion in 5 aspects: (1) the stage of tumor invasion, (2) the cellular origin of invasive lesions, (3) the significance of immunoreactive cells, (4) the significance of stromal cells, and (5) the potential approaches for interventions and prevention of tumor invasion. Our studies of breast tumors have obtained similar results and conclusions [65-74]. Our new hypothesis is the expansion of our previous one with the following new points of views:
a. the preservation of large clusters of genetically damaged stem or progenitor cells: Our previous hypothesis proposes that cell clusters overlying FBCLD represent tumor stem or progenitor cells, which undergo a series of immunohistochemical and morphologic changes, and finally transform into invasive lesions [43]. Our current hypothesis suggests that it is also possible that multiple genetically damaged primitive stem or progenitor cells within the same site may form large duct or acinar clusters that harbor the same genetic defects. These clusters may be formed immediately after external or internal insults during the early stage of morphogenesis, and progress rapidly, leading to early onset of prostate cancer at young ages. These clusters could also become maturation arrested at patients' early ages, while they retain the potential for unlimited proliferation and multi-lineage differentiation, representing “bad seeds for bad crops” at later ages.
b. direct transformation of the entire duct or acinar cluster into invasive lesions: Our previous hypothesis proposes that invasive cells are derived exclusively or preferentially from monoclonal proliferation of stem or progenitor cells overlying FBCLD [43]. Our current hypothesis suggests that, in addition to monoclonal proliferation, it is possible that the entire duct or acinar cluster may directly transform into invasive lesions after the disappearance of all surrounding basal cells and the basement membrane.
c. angiogenesis by genetically altered tumor stem cells: Our previous hypothesis proposes that a subset of luminal cell clusters overlying FBCLD are in direct physical continuity with vascular- or lymphatic duct-like structures, which allows them to progress directly to metastasis [43]. Our current hypothesis further suggests that some normal appearing duct or acinar clusters may retain genetically damaged primitive stem cells that could manufacture their own blood vessels or lymphatic ducts, which directly lead to metastasis.
The Significance of Our Hypothesis
Our hypothesis, if confirmed, could have several significant implications. Scientifically, it could lead to a new direction to explore novel approaches for early detection, intervention, and prevention of prostate tumor invasion. For example, as non-disrupted basal cell layers have significant inhibitory functions on epithelial cell growth, the development of therapeutic agents to stimulate basal cell growth or regeneration may provide a more effective approach for treatment and prevention of prostate cancer invasion. Clinically, it could potentially bring the following benefits:
- Better recognition of these clusters may avoid misdiagnosis and facilitate early interventions, which may significantly improve prognosis.
- Double immunohistochemical staining to assess the physical integrity of the basal cell layer, or an quantitative measurement of basal cell degeneration-related molecules in the blood or biopsies, may facilitate early identification of individuals at greater risk to develop invasive lesions.
- As genetic alterations not only define the scope and extent of, but also precede, both biochemical and morphological alterations, elucidation of the genetic profile of these normal appearing duct or acinar clusters may lead to the identification of the specific molecules that trigger the initiation of prostate carcinogenesis, progression, and invasion.
- Our technical approaches of assessing the physical and functional status of the basal cells may be used as a more reliable alternative for repeat biopsy to monitor prostate tumor progression and invasion.
More importantly, our hypothesis may be also applicable to progression and invasion of other epithelium derived tumors.
Acknowledgements
This study was supported in part by grants DAMD17-01-1-0129, DAMD17-01-1-0130, PC051308 from the US Congressionally Directed Medical Research Programs, grant BCTR0706983 from The Susan G. Komen Breast Cancer Foundation, grant 05AA from the AFIP/ARP joint research initiative project to Dr. Yan-gao Man, and grant 2006CB910505 from the Ministry of Chinese Science and Technology Department to Drs. Zhang Xichen, Yan-gao Man, and Gui-yuan Li.
The opinions and assertions contained herein represent the personal views of the authors and are not to be construed as official or as representing the views of the Department of the Army or the Department of Defense.
Conflict of Interest
The authors have declared that no conflict of interest exists.
References
1. Carruba G, Stefano R, Cocciaeliferro L. Intercellular communication and human prostate carcinogenesis. Ann NY Acad Sci. 2002;963:156-68
2. Goldstein NS, Underhiel J, Roszka N, Neill JS. Cytokeratin 34 beta E-12 immunoreactivity in benign prostate acini. Quantitation, pattern assessment, and electron microscopic study. Am J Clin Pathol. 1999;112:69-74
3. Bonkhoff H, Wernert N, Dhom G, Remberger K. Basement membranes in fetal, adult normal, hyperplastic and neoplastic human prostate. Virchows Arch A Pathol Anat Histopathol. 1991;418:375-81
4. Kosir MA, Wang W, Zukowski KL, Tromp G. Degradation of basement membrane by prostate tumor heparanase. J Surg Res. 1999;81:42-7
5. Bonkhoff H, Remberger K. Morphogenesis of benign prostatic hyperplasia and prostatic carcinoma. Pathology. 1998;19:12-20
6. Bostwick DG. Prospective origins of prostate carcinoma. Prostate intraepithelial neoplasia and atypical adenomatous hyperplasia. Cancer. 1996;78:330-6
7. Haggman MJ, Macoska JA, Wojno KJ, Oesterling JE. The relationship between prostate intraepithelial neoplasia and prostate cancer: critical issues. J Urol. 1997;58:12-22
8. Bonkhoff H, Remberger K. Differentiation pathways and histogenetic aspects of normal and abnormal prostatic growth: a stem cell model. Prostate. 1996;28:98-106
9. Barsky SH, Siegal GP, Jannotta F, Liotta LA. Loss of basement membrane components by invasive tumors but not by the benign counterparts. Lab Invest. 1983;49:140-7
10. Goldfarb RH, Liotta LA. Proteolytic enzymes in cancer invasion and metastasis. Semin Thromb Hemost. 1986;12:294-307
11. Gardner WA, Culberson DE. Atrophy and proliferation in the young adult Prostate. J Urol. 1987;137(1):53-6
12. Gardner WA. Hypothesis: Pediatric Origins of Prostate Cancer. Hum Path. 1995;26:1291-2
13. Bennett BD, Gardner WA. Embryonal hyperplasia of the prostate. Prostate. 1985;3(7):411-7
14. Malins DC, Polissar NL, Su Y, Gardner HS, Gunselman SJ. A new structural analysis of DNA using statistical models of infrared sepctra. Nat Med. 1997;3:927-30
15. Malins DC, Johnson PM, Barker EA. et al. Cancer-related changes in prostate DNA as men age and early identification of metastasis in prostate tumors. Proc Natl Acad Sci USA. 2003;100:5401-6
16. Malins DC, Anderson KM, Gilman NK. et al. Development of a cancer DNA phenotype prior to tumor formation. Proc Natl Acad Sci Sci USA. 2004;101:10721-5
17. Malins DC, Gilman NK, Green VM. et al. A DNA phenotype in healthy prostates, conserved in tumors and adjacent normal cells, implies a relationship to carcinogenesis. Proc Natl Acad Sci USA. 2005;102:19093-6
18. Ashida S, Nakagawa H, Katagiri T, Furihata M, Liizumi M, Anazawa Y. et al. Molecular features of the transition from prostate intraepithelial neoplasia (PIN) to prostate cancer: genome-wide gene-expression profiles of prostate cancers and PINs. Cancer Res. 2004;64:5963-72
19. Dawkins HJ, Sellner LN, Turbett GR, Thompson CA, Redmond SL, MeNeal JE, Cohen RJ. Distinction between intraductal carcinoma of the prostate (IDC-P), high-grade dysplasia (PIN), and invasive prostatic adecarcinoma, using molecular markers of cancer progression. Prostate. 2000;44:265-70
20. Harvei S, Skijorten FJ, Robsahm TE, Berner A, Tretli S. Is prostaticibtraepithelial neoplasi in the transitio/central zone a true presursor of cancer? A long-tern retrospectivestudy in Norway. Br J Cancer. 1998;78:46-9
21. Goeman L, Joniau S, Ponette D, Van der Aa F, Roskams T, Oyen R, Van Poppel H. Is low-grade prostatic intraepithelial neoplasia a risk factor for cancer. Prostate Cancer Prostatic Dis. 2003;6:305-10
22. Mostofi FK, Sesterhenn IA, Davis CJ Jr. Prostatic intraepithelial neoplasia (PIN): morphological clinical significance. Prostate Suppl. 1992;4:71-7
23. Kasahara Y, Tsukada Y. New insights and future advances in cancer diagnostics: Limitations of conventional tumor markers. In: (ed.) Nakarnura RM, Grody WW, Wu JT, Nagle RB. Cancer Diagnostics: Current and future trends. Totowa, NJ: Humanna Press. 2004
24. Joniau S, Goeman L, Pennings J, Van Poppel H. Prostatic intraepithelial neoplasia (PIN): importance and clinical managment. Eur Urol. 2005;48:379-85
25. Haggman MJ, Adolfsson J, Khoury S, Montie JE, Norlen J. Clinical managment of premalignant lesions of the prostate. WHO Collaborative Project and Consensus Conference on Public Health and Clinical Significance of Premalignant Alterations in the Genitourinary Tract. Scand J Urol Nephol Suppl. 2000;205:44-9
26. Parker SL, Tong T, Bolders S, Wingo PA. Cancer statistics. Cancer J Clin. 1997;47:5-27
27. Bostwick DG. Prostatic intraepithelial neoplasia is a risk factor for cancer. Semin Urol Oncol. 1999;17:187-9
28. Bostwick DG, Qian J, Frankel K. The incidence of high grade prostatic intraepithelial neoplasia in needle biopsies. J Urol. 1985;154:1791-4
29. Signoretti S, Waltregny D, Dilks J. et al. p63 is a prostate basal cell marker and is required for prostate development. Am J Pathol. 2000;157:1769-75
30. Kurita T, Medina RT, Mills AA, Cunha GR. Role of p63 and basal cells in prostate. Development. 2004;131:4955-64
31. Zou Z, Zhang W, Young D. et al. Maspin expression profile in human prostate cancer (caP) and in vitro induction of maspin expression by androgen ablation. Clin Cancer Res. 2002;8(5):1172-7
32. Cher ML, Biliran HRJr, Bhangat S. et al. Maspin expression inhibits osteolysis, tumor growth, and angiogenesis in animal model of prostate cancer bone metastasis. Proc Natl Acad Sci USA. 2003;100(13):7847-52
33. Man YG, Shen T, Zhao YG, Sang QXA. Focal prostate basal cell layer disruptions and leukocyte infiltration are correlated events: A potential mechanism for basal cell layer degradations and tumor invasion. Cancer Detect Prev. 2005;29:161-9
34. Man YG, Zhao CQ, Wang J, XL Chen. A subset of prostate basal cells lacks corresponding phenotypic markers. Pathology-Research & Practice. 2006;202(9):651-62
35. Man YG, Shen T, Zhao YG, Sang QX. Morphologically comparable prostate acini and ducts with and without a focal basal cell layer disruption have a different cell proliferation rate: Implications for tumor invasion. FASEB. 2004;18(5):A1193
36. Man YG, Mannion C, Shen T. Localized basal cell degeneration and resultant immunoreactions are a triggering factor for prostate tumor invasion. FASEB. 2005;19:A1517-8
37. Man YG, Shen T, Zhao YG, Sang QX. Focal prostate basal cell layer disruptions and leukocyte infiltration are correlated events: Implications for basal call layer degradation and tumor invasion. Cancer Detection & Prevention. 2004(S-51):15
38. Zhao C, Mannion C, Man YG. Potential roles of cytotoxic T-lymphocytes and natural killer cells in prostate basal cell layer disruptions and tumor invasion. Mod Pathol. 2005;18(175A):809
39. Man YG, Chen XL, Garcia FU, Gardner WA. Reduced p63 expression and elevated apoptosis in focally disrupted prostate basal cell layers: Implications for tumor invasion. Lab Investigation. 2006;86(292A):1358
40. Man YG, Liu XF, Mason J, Prabhakar S, Wang B, Zeng X, Stamatakos, Gardner WA. Prostate tumor cells near and distant from focally disrupted basal cell layers have different gene expression profiles. The American Society for Cell Biology 46th Annual Meeting. 2006;143:1843
41. Man YG, Liu AJ, Gardner WA. Normal appearing prostate acinar and duct clusters with malignant profiles. Mod Pathol. 2008;21(169A):770
42. Man YG, Liu AJ, Gardner WA. Elevated tenascin expression in stroma near focally disrupted prostate basal cell layers: implications for tumor progression and invasion. Mod Pathol. 2008;21(169A):771
43. Man YG, Gardner WA. Focal degeneration of basal cells and the resultant auto-immunoreactions: a novel mechanism for prostate tumor progression and invasion. Med Hypoth. 2008;70:387-408
44. Schwartz AM; Man YG, Rezaei MK, Simmens S, Berg PE. BP1. A homeoprotein, is significantly expressed in prostate adenocarcinomaand is concordant with prostatic intraepithelial neoplasia (PIN). Mod Pathol. accepted
45. Verona EV, Elkahloum AG, Yang J, Bandyopadhyay A, Yeh IT, Sun LZ. Transforming growth factor-beta signaling in prostate stromal cells supports prostate carcinoma growth by up-regulating stromal genes related to tissue remodeling. Cancer Res. 2007;67(12):5737-46
46. Zhou W, Grandis JR, Wells A. STAT3 is required but not sufficient for EGF receptor-mediated migration and invasion of human prostate carcinoma cell lines. Br J Cancer. 2006;95(2):164-71
47. Chung IW, Baseman A, Assikis V, Zhau HE. Molecular insights into prostate cancer progression: the missing link of tumor microenvironment. J Urol. 2005;173(1):10-20
48. Boulikas T. Control of DNA replication by protein phosphorylation. Anticancer Res. 1994;14:2465-72
49. Boulikas T. Phosphorylation of transcription factors and control of the cell cycle. Crit Rev Eukaryot Gene Expr. 1995;5:1-77
50. Chakravarthy MV, Spangenhurg EE, Booth FW. Culture in low levels of oxygen enhances in vitro proliferation potential of satellite cells from old skeletal muscles. Cell Mol Life Sci. 2001;58:1150-8
51. Csete M, Walikonis J, Slawny N. et al. Oxygen-mediated regulation of skeletal muscle satellite cell proliferation and adipogrnesis in culture. J Cell Physical. 2001;189:189-96
52. Studer L, Csete M, Lee SH. Enhanced proliferation, survival, and dopaminergic differentiation of CNS precursors in lowed pxygen. J Neurosci. 2000;20:7377-83
53. Klos KS, Wyszomierski SL, Sun M. et al. c-erbB2 increases vascular endothelial growth factor protein synthesis via activation of mammalian target of rapamycin/p70S6K leading to increased angiogenesis and spontaneous metastasis of human breast cancer cells. Cancer Res. 2006;66(4):2028-37
54. Hendrix MJ, Seftor EA, Hess AR, Seftor RE. Vasculogenic mimicry and tumor-cell plasticity: lessons from melanoma. Nat Rev Cancer. 2003;3(6):411-21
55. Kang Y, Massague J. Epithelial-mesenchymal transition: twist in development and metastasis. Cell. 2004;118:277-9
56. Sato T, Sakai T, Noguchi Y, Takita M, Hirakawa S, Ito A. Tumor-stromal cell contact promotes invasion of human uterine cervical carcinoma cells by augmenting the expression and activation of stromal matrix metalloproteinases. Gynecol Oncol. 2004;92:47-56
57. Strizzi L, Bianco C, Normanno N. Epithelial mesenchymal transition is a characteristic of hyperplasias and tumors in mammary gland from MMTV-Criptol-1 transgenic mice. J Cell Physiol. 2004;201:266-76
58. Gardner WA. "Pathology Paradigms". In: (ed.) Coffey D, Bruchovsky N, Gardner W, Resnick M, Karr J. Current Concepts and Approaches to the Study of Prostate Cancer. NY: A R Liss. 1987
59. Smith CJ, Gardner WA Jr. Inflammation - Proliferation: Possible Relationships in the Prostate. In: (ed.) Coffey D, Bruchovsky N, Gardner W, Resnick M, Karr J. Current Concepts and Approaches to the Study of Prostate Cancer. NY: A R Liss. 1987
60. Nelson G, Culberson DE, Gardner WA. Intraprostatic Spermatozoa. Hum Path. 1988;19(1):119-20
61. Gardner WA, Bennett BD. The Prostate - Overview: Recent Insights and Speculations. WA. Gardner and RS Weinstein (Eds.) Pathology and Pathobiology of Urinary Bladder and Prostate. Williams and Wilkins, Baltimore, MD. 1992
62. Peek RM Jr, Mohla S, DuBois RN. Inflammation in the genesis and perpetuation of cancer: summary and recommendations from a national cancer institute-sponsored meeting. Cancer Res. 2005;65:8583-6
63. MacLennan GT, Eisenberg R, Fleshman RL. et al. The influence of chronic inflammation in prostatic carcinogenesis: a 5-year follow-up study. J Urol. 2006;176:1012-6
64. Sell S, Pierce GB. Maturation arrest of stem cell differentiation is a common pathway for the cellular origin of teratocarcinomas and epithelial cancers. Lab Invest. 1994;70:6-22
65. Man YG, Tai L, Barner R, Vang R, Saenger JS, Shekitka KM. et al. Cell clusters overlying focally disrupted mammary myoepithelial cell layers and adjacent cells within the same duct display different immunohistochemical and genetic features: implications for tumor progression and invasion. Breast Cancer Res. 2003;5:R231-41
66. Man YG, Sang QXA. The significance of focal myoepitehlial cell layer disruptions in breast tumor invasion: a paradigm shift from the “protease-centered” hypothesis. Exp Cell Res. 2004;301:103-18
67. Yousefi M, Mattu R, Gao C, Man YG. Mammary ducts with and without focal myoepithelial cell layer disruptions show a different frequency of white blood cell infiltration and growth pattern: Implications for tumor progression and invasion. AIMM. 2005;13:30-7
68. Man YG, Shen T, Zhao YG, Sang QX. Focal prostate basal cell layer disruptions and leukocyte infiltration are correlated events: A potential mechanism for basal cell layer disruptions and tumor invasion. Cancer Detect Prev. 2005;29:161-9
69. Man YG, Zhang Y, Shen T, Vinh TN, Zeng X, Tauler J, Mulshine JL, Strauss BL. cDNA expression profiling identifies elevated expressions of tumor progression and invasion related genes in cell clusters of in situ breast tumors. Breast Cancer Res Treat. 2005;89:199-208
70. Man YG, Shen T, Weisz J, Berg PE, Schwartz AM, Mulshine JL, Sang QXA, Nieburgs HE. A subset of in situ breast tumor cell clusters lacks expression of proliferation and progression related markers but shows signs of stromal and vascular invasion. Cancer Detect Prev. 2005;29:323-31
71. Man YG, Zhao CQ, Wang J. Breast tumor cell clusters and their budding derivatives show different immunohistochemical profiles during stromal invasion: implications for hormonal and drug therapies. Cancer Therapy. 2006;4:193-204
72. Man YG, Nieburgs HE. A subset of cell clusters with malignant features in morphologically normal and hyperplastic breast tissues. Cancer Detect Prev. 2006;30(3):239-47
73. Man YG. Focal degeneration of aged or injured myoepithelial cells and the resultant auto- immunoreactions are trigger factors for breast tumor invasion. Medical Hypotheses. 2007;69(6):1340-57
74. Man YG. Bad seeds produce bad crops: a single-stage process of breast carcinogenesis. Bioscience Hypotheses. In press
Author contact
![]() Correspondence to: Yan-gao Man, MD., PhD., Director of Gynecologic and Breast Research Laboratory, Department of Gynecologic and Breast Pathology, Armed Forces Institute of Pathology and American Registry of Pathology, 6825 16th Street, NW, Washington DC 20306-6000. Phone: 202-782-1612; Fax: 202-782-3939; E-mail: manosd.mil
Correspondence to: Yan-gao Man, MD., PhD., Director of Gynecologic and Breast Research Laboratory, Department of Gynecologic and Breast Pathology, Armed Forces Institute of Pathology and American Registry of Pathology, 6825 16th Street, NW, Washington DC 20306-6000. Phone: 202-782-1612; Fax: 202-782-3939; E-mail: manosd.mil
Received 2008-7-9
Accepted 2008-8-6
Published 2008-8-11