ISSN: 1449-2288International Journal of Biological Sciences

Int J Biol Sci 2009; 5(7):667-678. doi:10.7150/ijbs.5.667 This issue Cite
Research Paper
Azithromycin suppresses interleukin-12p40 expression in lipopolysaccharide and interferon-γ stimulated macrophages
Keiko Yamauchi, Yoko Shibata ![]() , Tomomi Kimura, Shuichi Abe, Sumito Inoue, Daisuke Osaka, Michiko Sato, Akira Igarashi, Isao Kubota
, Tomomi Kimura, Shuichi Abe, Sumito Inoue, Daisuke Osaka, Michiko Sato, Akira Igarashi, Isao Kubota
Department of Cardiology, Pulmonology, and Nephrology, Yamagata University School of Medicine, 2-2-2 Iida-Nishi Yamagata 990-9585, Japan
Abstract
Azithromycin (AZM), a 15-member macrolide antibiotic, possesses anti-inflammatory activity. Macrophages are important in innate and acquired immunity, and produce pro-inflammatory cytokines such as interleukin (IL)-12, which are composed of subunit p40 and p35. The key function of IL-12 is the induction and maintenance of T-helper-1 responses, which is associated with the pathogenesis of chronic inflammatory diseases. We investigated the effect of azithromycin on IL-12p40 production in macrophages after lipopolysaccharide (LPS)/interferon (IFN)-γ stimulation. RAW264.7 macrophage cell line was pre-treated with vehicle or AZM, followed by the stimulation with LPS/IFN-γ. We measured IL-12 production by RT-PCR and ELISA. IL-12 transcriptional regulation was assessed by electrophoretic mobility shift assay and reporter assay. Phosphorylation of activator protein (AP)-1 and interferon consensus sequence binding protein (ICSBP) was assessed by immunoprecipitation using phosphotyrosine antibody, and immunoblotting using specific antibodies against JunB and ICSBP. AZM reduced the induction of IL-12p40 by LPS/IFN-γ in a dose dependent manner. AZM inhibited the binding of AP-1, nuclear factor of activated T cells (NFAT), and ICSBP, to the DNA binding site in the IL-12p40 promoter. AZM also reduced LPS/IFN-γ-induced IL-12p40 promoter activity. Phosphorylation of JunB and ICSBP was inhibited by azithromycin-treatment in stimulated cells. In conclusion, AZM reduced IL-12p40 transcriptional activity by inhibiting the binding of AP-1, NFAT, and ICSBP to the promoter site. This may represent an important mechanism for regulating the anti-inflammatory effects of AZM in macrophages.
Keywords: azithromycin, macrophage, interleukin-12, activator protein-1, interferon consensus sequence binding protein.
Introduction
The macrolide antibiotics have been used for the treatment of chronic lung diseases. The low dose and long-term administration of erythromycin (EM) for patients with diffuse panbronchiolitis (DPB) has reportedly significant effects on the clinical conditions of those patients [1]. Similar therapeutic effects of another 14-member macrolide, clarithromycin (CAM), have been reported in DPB patients [2]. The administration of azithromycin (AZM), a 15-member macrolide, has also been observed to improve the clinical condition of patients with chronic lung diseases, such as DPB [3] and cystic fibrosis [4,5].
Although the precise mechanisms by which the macrolides benefit patients with chronic lung diseases are not fully understood, circumstantial evidence suggests that it is based on their anti-inflammatory activity, rather than their antibacterial activity. We have previously demonstrated that CAM modulates the immune system by reducing interleukin (IL)-8 production via the suppression of activator protein (AP)-1 activity in bronchial epithelial cells [6], and that the 14-member macrolides, EM and CAM, enhance IL-10 signaling via signal transducer and activator of transcription-3/ suppressor of cytokine signaling-3 pathway after stimulation with lipopolysaccharide (LPS) [7].
Alveolar macrophages (AMs) are resident immune cells with important roles in host defense and inflammation in the lung [8]. Invading pathogens are recognized via pathogen recognition receptors, such as the toll-like receptors (TLR), the scavenger receptor, and the mannose receptor, and are phagocytosed by AMs [9]. These macrophages produce pro-inflammatory cytokines and chemokines, which accumulate and activate other inflammatory cells, such as neutrophils and T lymphocytes [10]. Thus, AMs' production of these pro-inflammatory cytokines is thought to be tightly linked to the severity of lung inflammation.
In treatment of chronic inflammatory diseases, the suppression of excess inflammatory cytokines is a potent therapeutic target. Based on the above mentioned action, macrolide antibiotics are one of the candidates for the treatment of chronic inflammatory diseases. AZM also possesses anti-inflammatory activity; AZM has been shown to reduce the production of the pro-inflammatory cytokines IL-12 and IL-6, and increase the production of the anti-inflammatory cytokine IL-10 in macrophage cell lines after stimulation with a combination of LPS and interferon (IFN)-γ (LPS/IFN-γ) [11].
IL-12 is a heterodimeric cytokine produced by macrophages and dendritic cells. The key roles of IL-12 are to promote the differentiation of naive T cells into T-helper-1 (Th1) cells, and to induce a Th1 immune response to bacteria, or their cell wall components, such as LPS. Thus, IL-12 is an important cytokine for cell-mediated immunity and host defense [12]. Over-expression of IL-12 is associated with the pathogenesis of chronic inflammatory diseases, such as Crohn's disease, and rheumatoid arthritis [13].
IL-12p40 and p35 are encoded on separate genes that form the biologically active p70 heterodimer. The p40 is not only a component of IL-12, but also that of IL-23. The p40 is only induced in cells that produce bioactive IL-12, while p35 gene is constitutively expressed in many cell types. IL-12 production is strongly induced by intracellular bacteria and bacterial products. Therefore, the transcription of IL-12 is dependent on the activity of the p40 promoter. Elements of this promoter include the AP-1 binding site (from -81 to -75) [14], and the interferon consensus sequence binding protein (ICSBP)/nuclear factor of activated T-cells (NFAT) heterodimer binding site (from -68 to -54) [15], both of which are functionally important for p40 promoter activation following LPS/IFN-γ stimulation. Although ICSBP cannot bind to this promoter element by itself, ICSBP heterodimerized with NFAT can [15].
On this basis, we hypothesized that AZM suppresses IL-12 production by repressing the activity of the IL-12p40 promoter. We investigated IL-12 expression and IL-12 promoter activity in macrophage cell lines pretreated with AZM upon the stimulation of LPS/IFN-γ.
Materials and Methods
Cell growth and Cell viability
Effects of Azithromycin on cell proliferation and viability were evaluated with the alamarBlue assay (BioSource International, Camarillo, CA). RAW264.7 cells (ACTT number TIB-71) were cultured at 37 °C for 0 - 72 h in 96-well plastic plates at a density of 4,000 cells/well in 20 μl of alamerBlue dye solution in 180 μL DMEM in the presence or absence of AZM (Pfizer, Inc. New York, NY) (0.1, 1.0, 10, and 20 μg/mL). The optical density (OD) of the culture medium was measured at 595 nm with spectrophotometoric microtiter plate reader (Model 450; Bio-Rad Laboratories, Inc., Herculs, CA) at 0, 1, 4, 6, 8, 24, 36, 48, 60, and 70 h.
Pre-treatment with AZM and stimulation with LPS/IFN-γ
RAW264.7 cells were seeded onto 6 cm dishes at 1×106 cells/dish and were pre-treated with vehicle (absolute ethanol) and AZM at 0, 1, 10 and 20 μg/mL, for 24 h. In all experiments, the final concentration of ethanol in culture medium was 0.5%. Cells were stimulated with LPS (100 ng/mL; Sigma-Aldrich, UK, L9143, LPS from Escherichia coli O111:448, purified by phenol extraction) and IFN-γ (400 U/mL; R&D Systems, Minneapolis, MN, USA) for 3 h (for total RNA isolation, nuclear protein and cytoplasmic protein preparations), or 8 h (for supernatant collection). Control cells were not stimulated with LPS/IFN-γ.
Reverse transcription (RT)-PCR
Total RNA from cells was extracted by RNeasy Kit (Qiagen, Valencia, CA, USA). RT was conducted using SuperScript III reverse transcriptase (Invitrogen Corp., Carlsbad, CA, USA). cDNA was amplified by Taq DNA polymerase (Platinum Taq DNA polymerase, Invitrogen) with specific primers: for IL-12p40 (forward; GTG AAG CAC CAA ATT ACT CCG G, reverse; GCT TCA TCA TCT GCA AGT TCT TGG G, 30 cycle), for GAPDH (forward; ACTC CAC TCA CGG CAA ATT CAA CGG, reverse; AGG GGC GGA GAT GAT GAC CC, 25 cycle), and for β-actin (forward; GTG GGC CGC TCT AGG CAC CAA, reverse; CTC TTT GAT GTC CAC GCA CGA TTT C, 25 cycle) [16]. PCR products were electrophoresed in 1.5% agarose gels containing ethidium bromide and visualized digitally with a UV illuminator (ATTO Bioscience, Tokyo, Japan). Band intensities were semi-quantified using computer software (Lane Analyzer 3.0; ATTO Bioscience, Tokyo, Japan).
ELISA
IL-12/IL-23p40 concentrations were measured using a sandwich ELISA kit (R&D Systems, Minneapolis, MN, USA) according to the manufacturer's protocol. Briefly, 50 μL supernatants were added into flat-bottom 96-well microtiter plates pre-coated with antibody against p40, followed by incubation for 2 h at room temperature. Plates were washed four times, followed by the addition of 100 µl/well antibody against p40 conjugated with horseradish peroxidase. After 2 hours of incubation, plates were washed four times with wash buffer. Then, the substrate solution was added, and the plates were incubated for 30 min at room temperature. The reaction was terminated with stop solution. Optical densities of plates were read at 450 nm in a microplete reader (Bio-Rad Laboratories).
Electrophoretic mobility shift assays (EMSA)
Nuclear extracts of RAW264.7 cells, with or without pre-treatment with AZM, were obtained, as described previously [17]. The activities of AP-1 and NFAT/ICSBP were assessed by EMSA with consensus oligonucleotides of IL-12p40 (AP-1: GAG ACT AGT CAG TTT, and ICSBP/NFAT: TCA GTT TCT ACT TTG GGT TTC CAT CAG AA). We used an EMSA kit (LightShift Chemiluminescent, Pierce Biotechnology, Inc., Rockford, IL, USA) with biotin end-labeled DNA probes instead of radioactive probes. Nuclear protein (4 μg) was incubated with labeled probes for 20 min at room temperature. The mixture was electrophoresed on a polyacrylamide gel and transferred to a nylon membrane. The biotin end-labeled DNA was detected according to the manufacturer's protocol. Band specificity was determined with competition experiments using a molar excess of unlabeled consensus oligonucleotides of AP-1, or ICSBP/NFAT that were added to the nuclear extracts before the addition of labeled probes.
Plasmids and site-directed mutagenesis
The murine IL-12p40 promoter, from the -101 to +55 region, was amplified from genomic DNA isolated from the mouse tail by PCR using an upstream primer containing the KpnI restriction site (GCC GGG TAC CTT TCA GTG TTG CAA TTG AGA CTA GTC AGT T) and a downstream primer containing the XhoI site (ACT TCT CGA GTT GCT TTG CTG CGA GCT GCC TGG TCT GAT GT). This was inserted into the luciferase reporter vector pGL4 (Promega Corp., Madison, WI, USA). The 3-bp mutants of AP-1, ICSBP and NFAT were generated using a site-directed mutagenesis kit (QuickChange II Site-Directed Mutagenesis Kit; Stratagene, La Jolla, CA, USA) using oligonucleotide primers, AP-1 (forward; GTT GCA ATT GAG ACT CTG CAG TTT CTA CTT TGG, reverse; CCA AAG TAG AAA CTG CAG AGT CTC AAT TGC AAC), ICSBP (forward; GAG ACT AGT CAG TTG ACA CTT TGG GTT TCC A, reverse; TGG AAA CCC AAA GTG TCA ACT GAC TAG TCT C), and NFAT (forward; GTC AGT TTC TAC TTT GTA GTT CCA TCA GAA AGT TC, reverse; GAA CTT TCT GAT GGA AAT CCA AAG TAG AAA CTG AC).
Transfection and reporter gene assays
Reporter plasmids containing the murine IL-12p40 promoter (from -101 to +55) or mutated forms of AP-1, ICSBP and NFAT, were transfected into RAW264.7 cells by Nucleofector (Amaxa Inc., Walkersville, USA). RAW264.7 cells were co-transfected with 2 μg of the pGL4/IL-12p40 promoter vector and 0.1 μg of the phRL-TK vector (Promega Corp., Madison, WI, USA). Twenty-four hours after transfection, cells were treated with vehicle or AZM (1.0 and 20 μg/mL) for 24 h, and subsequently stimulated with LPS/IFN-γ for 24 h. Cells were lysed with lysis buffer, and luciferase activity was assayed using the Dual Luciferase Assay System (Promega Biosciences, San Luis Obispo, CA, USA). First, firefly luciferase activities were assessed by a luminometer (MiniLumat LB 9506, Berthold, Germany). Then, after the addition of Stop & Glo solution (Promega), Renilla luciferase activities (an internal control for transfection efficiency) were also measured by by a luminometer.
Western blotting for JunB, ICSBP and NFAT
After LPS/IFN-γ stimulation, cytoplasmic and nuclear proteins were extracted from the AZM-treated RAW264.7 cells using the NE-PER nuclear and cytoplasmic extraction reagents kit (Thermo Fisher Scientific, Rockford, IL, USA).
Twenty microgram of cytoplasmic protein or 5 μg of nuclear protein were blotted onto a PVDF membrane (Amersham, Piscataway, NJ, USA). The membranes were blocked with milk, and treated with JunB antibody (1:200 dilution; Santa Cruz Biotechnology, Santa Cruz, CA, USA), ICSBP antibody (1:200 dilution; Santa Cruz Biotechnology), or NFATc1 antibody (1:200 dilution; Santa Cruz Biotechnology), followed by horseradish peroxidase-conjugated, anti-rabbit IgG (1:3000 dilution; Santa Cruz Biotechnology). These proteins were detected using the ECL plus system (Amersham), and exposed on Fuji X-ray film.
Immunoprecipitation and immunoblotting
Nuclear proteins were extracted from RAW264.7 cells pre-treated with vehicle or AZM (1.0 and 20 μg/mL) followed by stimulation with LPS/IFN-γ for 3 h. Immunoprecipitation was performed with 200 μg nuclear protein. Nuclear proteins were diluted into RIPA buffer containing both protease and phosphatase inhibitors [18], and incubated with 1 μL of phosphotyrosine antibody (4G10, Millipore, Bedford, MA, USA) for 4 h at 4 °C, followed by incubation with 15 μL of 50% sepharose G protein beads for 1 h (Sigma-Aldrich, UK). Beads were washed three times with RIPA buffer (without SDS), and proteins were eluted by boiling in SDS sample buffer, separated on 12% SDS-PAGE, and transferred onto a nitrocellulose membrane. Blots were probed with antibody against JunB (1:200 dilution; Santa Cruz Biotechnology) or against ICSBP (1:200 dilution; Santa Cruz Biotechnology), and protein signals were detected with the ECL plus system (Amersham) for JunB, and Western Breeze (Invitrogen Corp., Carlsbad, CA, USA) for ICSBP.
Statistical analysis
Multiple comparisons were performed by one way ANOVA followed by the Student-Newman-Keuls method. Results are expressed as the mean ± SD. Statistical significance was taken to be P < 0.05. Statistical analyses were done using computer software (StatView 5.1, SAS Institute, Inc., Cary, NC, USA).
Results
Effects of AZM on the cell viability of RAW 264.7cells
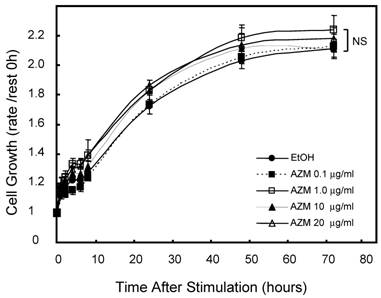
First, we assessed the influence of AZM on cell growth and viability by alamerBlue assay. Neither lower (0.1 and 1.0 μg/mL) nor higher (10 and 20 μg/mL) concentrations of AZM affected the proliferation and viability of RAW 264.7 cells (Figure 1). In microscopic observations, RAW 264.7 cells grew to up to 90-100% confluence in a 96-well culture plate after incubation for 72 h, no morphologic changes were observed in resting cells or AZM-treated cells. Nevertheless, the question remains whether there is broad-based inhibition of transcriptional activity because the cells are metabolically affected by the AZM treatment. We also examined the expression of β-actin, a cytoskeleton protein, which is widely used as internal control gene in RT-PCR. The expression levels of GAPDH/β-actin mRNAwere not changed by AZM treatment for 72 h (Relative expression: vehicle 1.00 ± 0.12, AZM 1.0 μg/mL 1.12 ± 0.24, and AZM 20 μg/mL 0.98 ± 0.10, p = 0.94, n = 5 in each group).
Effect of azithromycin (AZM) on RAW264.7cell growth and cell viability. Cells were cultured with alamarBlue dye solution in absence or presence of AZM (0.1, 1.0, 10, and 20 μg/ml) for 72 h, and the optical density (OD) at 595 nm was measured at the indicated time points. Proliferation and viability of RAW264.7 cells were determined in relation to growth curve, based on the measurement of OD at each time point. Data are mean value of six independent experiments.
AZM inhibited IL-12p40 gene expression induced by LPS/IFN-γ
IL-12p40 gene expression was not detected in the control cells (treated with vehicle only), or AZM treated RAW264.7 cells not stimulated with LPS/IFN-γ (Figure 2A). Cells stimulated with LPS/IFN-γ for 3 h showed elevated IL-12p40 gene expression by RT-PCR. The LPS/IFN-γ-induced IL-12p40 gene expression was inhibited by pre-treatment with AZM in a dose dependent manner (Figure 2A).
The production of IL-12p40 in RAW264.7 macrophage cells pre-treated with azithromycin (AZM) and stimulated with LPS/IFN-γ. (A) RAW264.7 cells were pre-treated with AZM (1.0 and 20 μg/mL) for 24 h, and stimulated with LPS/IFN-γ for 3 h. Gene expression of IL-12p40 was evaluated by RT-PCR. The expression of IL-12p40 gene was increased by LPS/IFN-γ stimulation compared with the control cells. This increased expression was inhibited by AZM in a dose dependent manner. (B) The p40 protein levels were measured by ELISA in the supernatants of cells pre-treated with vehicle, or AZM, for 24 h, followed by stimulation with LPS/IFN-γ for 8 h. The p40 protein levels were increased by LPS/IFN-γ, and these levels were significantly reduced by pre-treatment with AZM. Each group comprised five independent experiments. Data are expressed as the mean ± SD. # P < 0.05 vs. vehicle vehicle-pretreatment followed by LPS/IFN-γ stimulation, ## P < 0.01 vs. vehicle-pretreatment followed by LPS/IFN-γ stimulation. N.D. indicates that the expression was not detected.
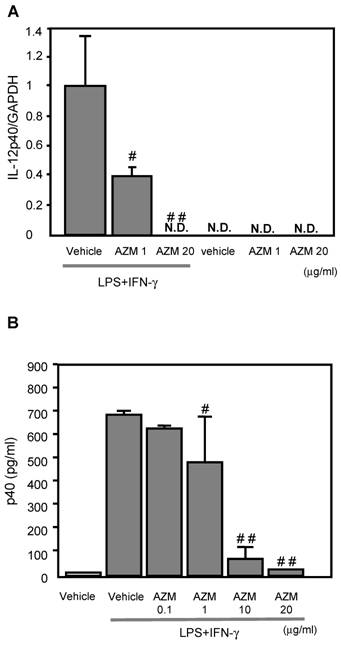
AZM reduced the production of p40 protein induced by LPS/IFN-γ
The levels of p40 protein in the supernatant from cells stimulated with LPS/IFN-γ for 8 h were significantly elevated, compared with those of unstimulated cells. The LPS/IFN-γ-induced p40 production was inhibited by pre-treatment with AZM in a dose dependent manner (Figure 2B).
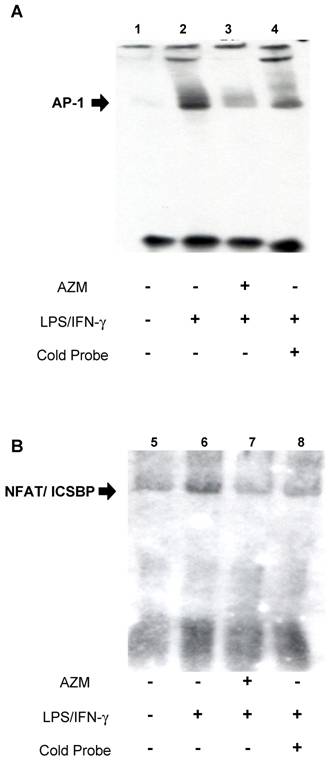
AZM inhibited interactions between transcription factors (AP-1, ICSBP, and NFAT) and IL-12p40 promoter elements
EMSA was used to assess the effect of AZM on DNA-protein interactions in the IL-12p40 promoter. Using the AP-1 probe, an increase in DNA-nuclear protein complexes was observed in RAW264.7 cells stimulated with LPS/IFN-γ (Figure 3A, lane 2), compared with unstimulated cells (Figure 3A, lane 1). This increase in DNA-protein complex was reduced by co-incubation with an excess of unlabeled AP-1 oligonucleotide (Figure 3A, lane 4), and by pre-treatment with AZM (Figure 3A, lane 3). Similarly, using the ICSBP/NFAT probe, an increase in DNA-nuclear protein complexes was observed in LPS/IFN-γ stimulated cells (Figure 3B, lane 6) compared with unstimulated cells (Figure 3B, lane 5), and this increase was inhibited by co-incubation with an excess of unlabeled ICSBP/NFAT oligonucleotide (Figure 3B, lane 8), and pre-treatment with AZM (Figure 3B, lane 7).
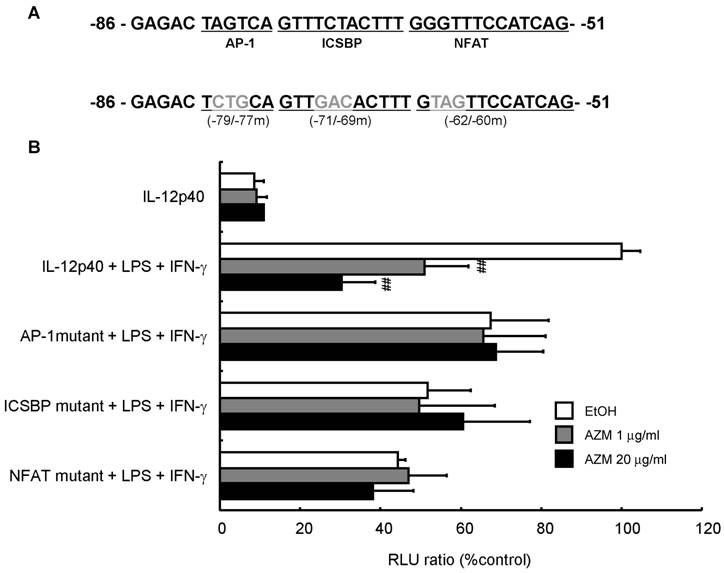
AZM reduced IL-12p40 promoter activity following LPS/IFN-γ stimulation
To investigate whether AZM affected promoter activity by changing the affinity between the binding elements in the IL-12p40 promoter and transcription factors, we performed an assay using a reporter plasmid with a 3-bp mutation in the AP-1 (-79 to -77), ICSBP (-71 to -69), or NFAT (-62 to -60) binding sites (Figure 4A). AZM decreased IL-12p40 reporter activity of LPS/IFN-γ treated cells in a dose dependent manner (Figure 4B). Mutation of the AP-1, ICSBP or NFAT sites in the IL-12p40 promoter resulted in decreased luciferase signals after stimulation with LPS/IFN-γ. Furthermore, these mutations abrogated the inhibitory effects of AZM on LPS/IFN-γ-induced promoter activities (Figure 4B).
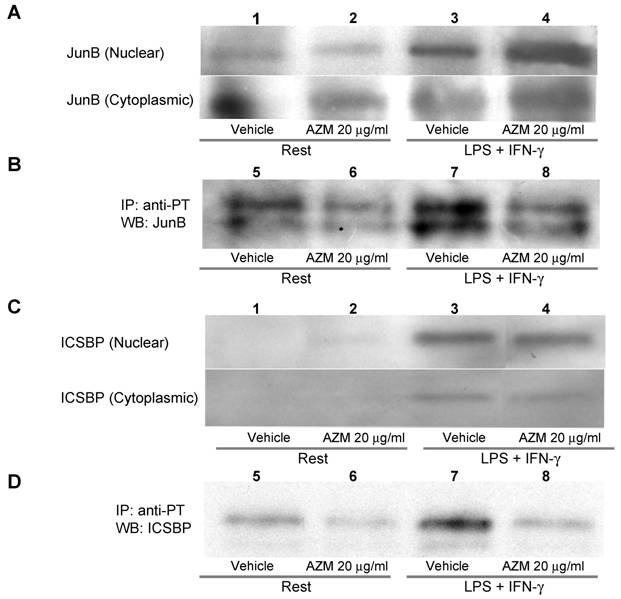
AZM inhibited phosphorylation of JunB and ICSBP
The AP-1 binding motif in the IL-12p40 promoter is reportedly associated with JunB, c-Jun or c-Fos [14]. Among these components of AP-1, phosphorylation of JunB is related to binding to DNA, and to the expression of AP-1 regulated genes [19]. Tyrosine phosphorylation of ICSBP reportedly increases the affinity of protein-DNA interactions [20]. Stimulation with LPS/IFN-γ elevated the protein levels of JunB and ICSBP in the nuclear fraction (Figure 5A and 5C, upper panel lane 3), and this increase was not affected by pre-treatment with AZM (Figure 5A and 5C, upper panel lane 4). Stimulation with LPS/IFN-γ elevated the levels of phosphorylated JunB and ICSBP in the nuclear fraction (Figure 5B and 5D, lane 7), and these elevated levels were decreased by pre-treatment with AZM (Figure 5B and 5D, lane 8).
NFAT levels were not increased after stimulation with LPS/IFN-γ (data not shown). Levels of nuclear NFAT in cells following LPS/IFN-γ stimulation were not increased compared with those of unstimulated cells (data not shown). AZM did not change the level of nuclear NFAT (data not shown).
Azithromycin (AZM) inhibited the association of activator protein (AP)-1 and interferon consensus sequence binding protein (ICSBP)/ nuclear factor of activated T-cells (NFAT) with DNA-binding sites in the IL-12p40 promoter. Effect of AZM on AP-1 binding activity for the IL-12p40 promoter was assessed by EMSA. Nuclear extracts were obtained from resting RAW264.7 cells (lane 1), cells pre-treated with vehicle followed by stimulation with LPS/IFN-γ (lane 2), and from cells pre-treated with AZM (20 μg/mL) followed by stimulation with LPS/IFN-γ (lane 3). In lane 4, nuclear extract from cells pre-treated with vehicle followed by stimulation with LPS/IFN-γ were treated with excess cold probe. AP-1-DNA binding activity was enhanced by LPS/IFN-γ stimulation. Enhanced binding was inhibited by pre-treatment with AZM. Specific competition with cold probe eliminated the DNA-nuclear protein complex band. Effect of AZM on ICSBP/NFAT binding activity for IL-12p40 promoter was assessed by EMSA. Nuclear extracts were obtained from resting RAW264.7 cells (lane 5), cells pre-treated with vehicle followed by stimulation with LPS/IFN-γ (lane 6), and from cells pre-treated with AZM (20 μg/mL) followed by stimulation with LPS/IFN-γ (lane 7). In lane 8, nuclear extract from cells pre-treated with vehicle followed by stimulation with LPS/IFN-γ were treated with excess cold probe. ICSBP/NFAT DNA binding activity was enhanced by LPS/IFN-γ stimulation. Enhanced binding was inhibited by pre-treatment with AZM. Specific competition with cold probe eliminated the DNA-nuclear protein complex band. Representative images from three independent experiments are shown. “+” indicates the presence of pretreatment of AZM, stimulation of LPS/IFN-γ or cold probe in experiment of each lane. “-” indicates the absence of pretreatment of AZM, stimulation of LPS/IFN-γ or cold probe in experiment of each lane.
Inhibition of IL12-p40 promoter activity by azithromycin (AZM) in RAW264.7 macrophage cells. (A) Three-base pair substitution mutants were created from -79 to -77 [activator protein (AP)-1 binding site], -71 to -69 [interferon consensus sequence binding protein (ICSBP) binding site], and -62 to -60 [nuclear factor of activated T-cells (NFAT) binding site], in the IL-12p40 promoter. (B) RAW264.7 cells were co-transfected with wild-type, or each mutant reporter plasmid, and phRL-TK reporter plasmid. After 24 h, cells were incubated with vehicle or AZM for 24 h, and stimulated with LPS/IFN-γ for 24 h. Luciferase activity was normalized to phRL-TK activity, and results expressed as a percentage of promoter activity compared with LPS/IFN-γ stimulation for wild-type -101 to +55 IL-12p40 promoter-luciferase plasmid (100%). Stimulation with LPS/IFN-γ markedly increased the luciferase activity in the wild-type IL-12p40 promoter, and AZM significantly reduced LPS/IFN-γ-induced IL-12p40 promoter activity in a dose dependent manner. Each mutation of AP-1, ICSBP, and NFAT repressed LPS/IFN-γ-induced promoter activity, and abrogated the inhibitory effects of AZM on IL-12p40 promoter activity. Five independent experiments in each group, ## P < 0.01 vs. wild-type + LPS +IFN-γ.
Azithromycin (AZM) inhibited phosphorylation of JunB and interferon consensus sequence binding protein (ICSBP). Nuclear and cytoplasmic fractional proteins were prepared from RAW264.7 cells stimulated with LPS/IFN-γ for 3 h in the presence of vehicle or AZM. Stimulation of LPS/IFN-γ elevated JunB and ICSBP protein levels in nuclear fractions (A and C, upper panel lane 3). However, AZM did not reduce the protein levels of JunB and ICSBP (A and C, upper panel lane 4). Nuclear proteins were immunoprecipitated with an antibody against phosphotyrosine. Immunoprecipitated proteins were immunoblotted with antibody against JunB and ICSBP. The phosphorylation of JunB and ICSBP was enhanced by stimulation of LPS/IFN-γ (B and D, lane 7). This enhanced phosphorylation was abrogated by AZM treatment (B and D, lane 8).
Discussion
We have shown that pre-treatment with AZM reduces the production of IL-12 in LPS/IFN-γ stimulated macrophages (Figure 2). Murphy et al. demonstrated that AZM suppressed IL-12 production in AMs [11], however, no mechanism was proposed. The present study demonstrates one of the mechanisms by which AZM suppresses IL-12 in macrophages.
IL-12 transcriptional regulation reportedly occurs via the IL-12p40 promoter through its interaction with transcription factors, such as AP-1, ICSBP and NFAT [14,15]. In the present study, AZM was shown to inhibit binding of these transcriptional factors to their DNA binding-sites (Figure 3). IL-12p40 promoter activity was inhibited by AZM in a dose dependent manner (Figure 4B). In addition, mutation of AP-1, ICSBP or NFAT in this promoter region abrogated the inhibitory effect of AZM (Figure 4B). We also showed that treating cells with AZM prior to stimulation with LPS/IFN-γ, reduced the levels of active (tyrosine phosphorylated) JunB and ICSBP produced (Figure 5B & 5D). It is likely that the inhibitory effect of AZM on the phosphorylation of JunB and ICSBP is one of the key mechanisms in the suppression of IL-12 production by macrophages.
Although there is no detection of IL-12p40 gene expression in vehicle-treated cells, basal levels of p40 protein are detected in the supernatant (Figure 2). This means that the gene expression of IL-12p40 in untreated cells is under the detection limit in this PCR system. As shown in Figure 5B & 5D, vehicle-treated cells exhibit detectable levels of active, tyrosine-phosphorylated JunB and ICSBP, but the levels of tyrosine-phosphorylated JunB and ICSBP in unstimulated cells were significantly low, compared with those of LPS/IFN stimulated cells (Figure 5B & 5D). These phosphorylated JunB and ICSBP in unstimulated cells may be implicated in the detectable level of IL-12p40 protein in unstimulated cells.
NFAT usually exists in phosphorylated form, and resides in the cytoplasm in the rest condition. Upon the stimulation, NFAT is dephosphorylated, and is translocated into the nucleus [21]. In the present study, the nuclear protein level of NFAT in cells stimulated with LPS/IFN-γ was not changed by AZM. Because the heterodimerized complex of ICSBP/NFAT reportedly binds to the IL-12p40 promoter and activates it [15], a reduction in the level of NFAT is not necessarily associated with the reduction of IL-12 p40 promoter activity by AZM in LPS/IFN-γ stimulated cells.
The mechanism for repressing the phosphorylation of JunB and ICSBP is still unclear. We and other investigators have previously demonstrated that 14-member macrolides enhanced the production of anti-inflammatory cytokine IL-10 [7,11]. It is reported that IL-10 suppresses the phosphorylation of AP-1 [22]. Taken together, it may be possible that AZM enhances the production of IL-10, which suppresses the phosphorylation of JunB and ICSBP.
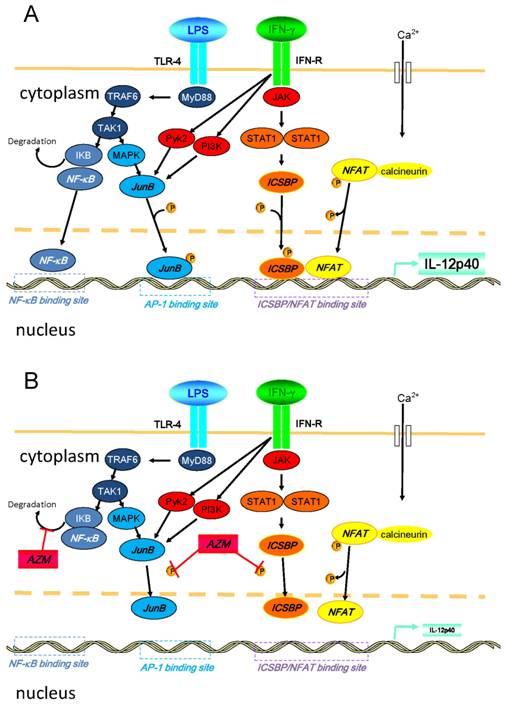
Another possible explanation for this suppression of IL-12 is repression of nuclear factor (NF)-κB pathway that is a key transcription factor related to the expression of pro-inflammatory cytokines. In our preliminary experiment, we examined the expression of TLR4 in the AZM-pretreated cells upon the stimulation of LPS/IFN-γ, and observed that the expression of TLR4 remained unchanged (data not shown). However, it has already been reported that AZM suppressed NF-κB pathway [23]. Thus, it is likely that the modulation of NF-κB signaling is involved in the mechanisms for the suppression of IL-12 by AZM. In Figure 6, we summarized the intracellular pathways that lead to the production of IL-12p40 and the mechanisms of AZM to inhibit the expression of IL-12p40 [14,15,23-26].
Diagrammatic representation of intracellular pathways that lead to the production of IL-12p40 and the mechanisms of azithromycin (AZM) to inhibit the expression of IL-12p40 upon the stimulation of LPS/IFN-γ. (A) Inflammatory stimulation leads to the activation of various signal transduction cascades in cells. Binding of LPS and toll-like receptor (TLR)-4 activates NF-κB. Soon after occurring LPS/TLR-4 signaling, IKB is degraded in this process. Subsequently, NF-κB translocates into the nucleus, and binds to its binding motif in promoter region of IL-12p40. Moreover, LPS/TLR-4 signaling results in the activation of MAPK pathway, which leads to the phosphorylation of activator protein (AP)-1, such as JunB. Phosphorylated AP-1 binds to its binding motif in the promoter. Binding of IFN-γ and its receptor activates JAK-STAT pathways, and other pathways, such as PI3K and Pyk2. Activated STAT1 phosphorylates ICSBP. In this cellular activation process, calcium influx is induced, and elevated intracellular concentration of calcium activates calcineurin. Then, NFAT is dephosphorylated by activated calcineurin, and subsequently translocates into the nucleus. NFAT heterodimerizes with ICSBP, and this heterodimer binds to the binding motif in promoter. Activated PI3K and Pyk2 also activate the MAPK pathway. (B) In the presence of AZM, the phosphorylation of ICSBP and JunB is inhibited. This inhibition results in the reduced binding capacity of ICSBP/NFAT and JunB to their DNA binding motif in the IL-12p40 promoter, and IL-12p40 gene expression is suppressed. It was also reported that AZM inhibits NF-κB pathway by reducing the degradation of IKB (reference 23). Abbreviations: ICSBP, interferon consensus sequence binding protein; IKB, inhibitor of κB; JAK, Janus kinase; MAPK, mitogen activated protein kinase; MyD88, myeloid differentiation primary response gene 88; NFAT, nuclear factor of activated T-cells; NF-κB, nuclear factor-κB; PI3K, phosphatidylinositol 3-kinase, Pyk2, calcium-dependent tyrosine kinase; STAT1, signal transducer and activator of transcription 1; TAK1, transforming growth factor-beta-activated kinase 1; TRA6, TNF receptor-associated factor 6.
IL-12 plays an important role in promoting a Th1 response while suppressing a Th2 response. IL-12-deficient mice and IL-12 neutralizing antibodies reportedly suppress Th1 responses, and reduce protection against intracellular pathogens [12]. IL-12 knockout mice exhibit limited inflammatory reactions [27], whereas IL-12 p40 over-expression is related to chronic inflammatory and autoimmune diseases [13,28]. Th1 cytokine-induced inflammation is associated with chronic lung diseases. The levels of IL-12 in exhaled breath condensate of COPD patients are elevated, compared with those of control subjects [29]. In addition, CD8 positive T lymphocytes, Th1 cytokine-producing cells, are increased in the lung of COPD patients [30,31]. Taken together, the balance of Th1/Th2 balance in the lung of COPD patients seems to be shifted toward Th1 dominant. Furthermore, Th1 cytokines are increased in the lung of DPB patients [32].
Accumulating evidences suggest that use of macrolides has many beneficial effects on such chronic lung diseases. Seemungal and colleagues reported that administration of long-term erythromycin (250 mg/day) reduced the exacerbation in COPD patients [33]. Clarithromycin prevents the development of pulmonary emphysema in cigarette smoking mice [34]. Long-term and low dose administration of 14-member macrolide antibiotics is an effective treatment for DPB [1,2].
Cystic fibrosis is an autosomal recessive disease primarily affecting the lungs. To date, any curative treatment for this disease has not been established. The pathophysiology of cystic fibrosis lung disease is related to the abnormal cystic fibrosis transmembrane receptor present in the apical cell membrane of airway epithelial cells [35]. Dysfunction of bronchial epithelial cells creates an inflammatory milieu in airways, causing damage to the airway system [35]. Because chronic airway inflammation is deeply involved in the pathophysiology of cystic fibrosis, AZM is hoped to be as a part of the standard maintenance therapy against this disease [35]. In fact, the administration of AZM to the murine model of cystic fibrosis attenuates the accumulation of AMs after LPS-induced inflammation [36].
In these chronic lung diseases, excess pro-inflammatory cytokines play harmful roles for the deterioration of disease severity. Therefore, the attenuation of excess pro-inflammatory cytokines is a potent therapeutic target for such diseases. Indeed, pro-inflammatory cytokines, such as TNF-α, IL-1β, IL-6 and IL-8, are known to be suppressed by AZM [23,37]. Because IL-12 plays key roles in Th1-related diseases, the attenuation of IL-12 production by AZM may also contribute to improve the condition of chronic lung diseases. Furthermore, because AZM inhibits the production of IL-12p40 protein, it is suggested that AZM also attenuates the expression of IL-23, the cytokine composed of IL-12p40 and IL-23p19 [38]. IL-23 plays key roles in autoimmune diseases and graft rejection through the activation of Th-17 cells [38]. Therefore, AZM might have some beneficial effects on autoimmune diseases and graft rejection.
Furthermore, the colonization of bacteria or fungus is frequently observed in chronic airway diseases. Thus, excess immune suppression is risk for the growth of these pathogens. Corticosteroid and immune suppressants, such as cyclosporine, cause strong immune suppression. Long term administration of these medicines sometimes causes respiratory infection, such as pneumonia. However, immune modulation of macrolide antibiotics is not so strong that, to our best knowledge, long term macrolide therapy does not result in the deterioration of infections.
Macrolide antibiotics are suitable in the treatment of respiratory tract infections due to their good penetration into lung tissue. Levels of AZM after oral administration of 500 mg reach 2.18 μg/mL in epithelial lining fluid, and 23.0 μg/mL in AMs [39]. Thus, the dose of AZM used in this study was comparable with that achieved therapeutically.
In conclusion, AZM reduced IL-12 p40 transcriptional activity by inhibiting the binding of AP-1, NFAT, and ICSBP to this promoter site. This may represent a mechanism for the regulation of the anti-inflammatory effects of AZM in macrophages.
Acknowledgements
This study was supported by a Grant-in-aid from the Global COE program of the Japan Society for the Promotion of Science and grants-in-aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology, Japan (18590835, 18790530, and 19590880).
Conflict of Interest
All authors do not have a financial relationship with a commercial entity that has an interest in the subject of this manuscript.
References
1. Kudoh S, Azuma A, Yamamoto M. et al. Improvement of survival in patients with diffuse panbronchiolitis treated with low-dose erythromycin. Am J Respir Crit Care Med. 1998;157:1829-32
2. Kadota J, Mukae H, Ishii H. et al. Long-term efficacy and safety of clarithromycin treatment in patients with diffuse panbronchiolitis. Respir Med. 2003;97:844-50
3. Tamaoki J, Kadota J, Takizawa H. Clinical implications of the immunomodulatory effects of macrolides. Am J Med. 2004 117 Suppl 9A: 5S-11S
4. Clement A, Tamalet A, Leroux E. et al. Long term effects of azithromycin in patients with cystic fibrosis: A double blind, placebo controlled trial. Thorax. 2006;61:895-902
5. Carr RR, Nahata MC. Azithromycin for improving pulmonary function in cystic fibrosis. Ann Pharmacother. 2004;38:1520-4
6. Abe S, Nakamura H, Inoue S. et al. Interleukin-8 gene repression by clarithromycin is mediated by the activator protein-1 binding site in human bronchial epithelial cells. Am J Respir Cell Mol Biol. 2000;22:51-60
7. Yamauchi K. Enhanced interleukin-10 signaling with 14-member macrolides in lipopolysaccharides-stimulated macrophages. EXCLI J. 2008;7:169-76
8. Shibata Y, Berclaz PY, Chroneos ZC. et al. GM-CSF regulates alveolar macrophage differentiation and innate immunity in the lung through PU.1. Immunity. 2001;15:557-67
9. Akira S. Toll-like receptor signaling. J Biol Chem. 2003;278:38105-8
10. Plowden J, Renshaw-Hoelscher M, Engleman C. et al. Innate immunity in aging: Impact on macrophage function. Aging Cell. 2004;3:161-7
11. Murphy BS, Sundareshan V, Cory TJ. et al. Azithromycin alters macrophage phenotype. J Antimicrob Chemother. 2008;61:554-60
12. Trinchieri G Interleukin-12. A proinflammatory cytokine with immunoregulatory functions that bridge innate resistance and antigen-specific adaptive immunity. Annu Rev Immunol. 1995;13:251-76
13. Seder RA, Kelsall BL, Jankovic D. Differential roles for IL-12 in the maintenance of immune responses in infectious versus autoimmune disease. J Immunol. 1996;157:2745-8
14. Zhu C, Gagnidze K, Gemberling JH. et al. Characterization of an activation protein-1-binding site in the murine interleukin-12 p40 promoter. Demonstration of novel functional elements by a reductionist approach. J Biol Chem. 2001;276:18519-28
15. Zhu C, Rao K, Xiong H. et al. Activation of the murine interleukin-12 p40 promoter by functional interactions between NFAT and ICSBP. J Biol Chem. 2003;278:39372-82
16. Scharton-Kersten T, Contursi C, Masumi A. et al. Interferon consensus sequence binding protein-deficient mice display impaired resistance to intracellular infection due to a primary defect in interleukin 12 p40 induction. J Exp Med. 1997;186:1523-34
17. Yoshida M, Korfhagen TR, Whitsett JA. Surfactant protein D regulates NF-kappa B and matrix metalloproteinase production in alveolar macrophages via oxidant-sensitive pathways. J Immunol. 2001;166:7514-9
18. Dignam JD, Lebovitz RM, Roeder RG. Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res. 1983;11:1475-89
19. Hulboy DL, Matrisian LM, Crawford HC. Loss of JunB activity enhances stromelysin 1 expression in a model of the epithelial-to-mesenchymal transition of mouse skin tumors. Mol Cell Biol. 2001;21:5478-87
20. Sharf R, Meraro D, Azriel A. et al. Phosphorylation events modulate the ability of interferon consensus sequence binding protein to interact with interferon regulatory factors and to bind DNA. J Biol Chem. 1997;272:9785-92
21. Shaw KT, Ho AM, Raghavan A. et al. Immunosuppressive drugs prevent a rapid dephosphorylation of transcription factor NFAT1 in stimulated immune cells. Proc Natl Acad Sci U S A. 1995;92:11205-9
22. Mohamed SG, Sugiyama E, Shinoda K. et al. Interleukin-10 inhibits RANKL-mediated expression of NFATc1 in part via suppression of c-Fos and c-Jun in RAW264.7 cells and mouse bone marrow cells. Bone. 2007;41:592-602
23. Aghai ZH, Kode A, Saslow JG. et al. Azithromycin suppresses activation of nuclear factor-kappa B and synthesis of pro-inflammatory cytokines in tracheal aspirate cells from premature infants. Pediatr Res. 2007;62:483-8
24. Gough DJ, Levy DE, Johnstone RW. et al. IFNgamma signaling-does it mean JAK-STAT?. Cytokine Growth Factor Rev. 2008;19:383-94
25. Lu YC, Yeh WC, Ohashi PS. LPS/TLR4 signal transduction pathway. Cytokine. 2008;42:145-51
26. Zanoni I, Ostuni R, Capuano G. et al. CD14 regulates the dendritic cell life cycle after LPS exposure through NFAT activation. Nature. 2009;460:264-8
27. Huaux F, Arras M, Tomasi D. et al. A profibrotic function of IL-12p40 in experimental pulmonary fibrosis. J Immunol. 2002;169:2653-61
28. Adorini L. Interleukin-12, a key cytokine in Th1-mediated autoimmune diseases. Cell Mol Life Sci. 1999;55:1610-25
29. Dentener MA, Creutzberg EC, Pennings HJ. et al. Effect of infliximab on local and systemic inflammation in chronic obstructive pulmonary disease: A pilot study. Respiration. 2008;76:275-82
30. Hodge G, Nairn J, Holmes M. et al. Increased intracellular T helper 1 proinflammatory cytokine production in peripheral blood, bronchoalveolar lavage and intraepithelial T cells of COPD subjects. Clin Exp Immunol. 2007;150:22-9
31. Saetta M, Mariani M, Panina-Bordignon P. et al. Increased expression of the chemokine receptor CXCR3 and its ligand CXCL10 in peripheral airways of smokers with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2002;165:1404-9
32. Park SJ, Lee YC, Rhee YK. et al. The effect of long-term treatment with erythromycin on Th1 and Th2 cytokines in diffuse panbronchiolitis. Biochem Biophys Res Commun. 2004;324:114-7
33. Seemungal TA, Wilkinson TM, Hurst JR. et al. Long-term erythromycin therapy is associated with decreased chronic obstructive pulmonary disease exacerbations. Am J Respir Crit Care Med. 2008;178:1139-47
34. Nakanishi Y, Kobayashi D, Asano Y. et al. Clarithromycin prevents smoke-induced emphysema in mice. Am J Respir Crit Care Med. 2009;179:271-8
35. Florescu DF, Murphy PJ, Kalil AC. Effects of prolonged use of azithromycin in patients with cystic fibrosis: A meta-analysis. Pulm Pharmacol Ther. 2009: [Epub ahead of print].
36. Legssyer R, Huaux F, Lebacq J. et al. Azithromycin reduces spontaneous and induced inflammation in deltaF508 cystic fibrosis mice. Respir Res. 2006;7:134
37. Meyer M, Huaux F, Gavilanes X. et al. Azithromycin reduces exaggerated cytokine production by M1 alveolar macrophages in cystic fibrosis. Am J Respir Cell Mol Biol. 2009 [Epub ahead of print]
38. Kikly K, Liu L, Na S. et al. The IL-23/Th17 axis: Therapeutic targets for autoimmune inflammation. Curr Opin Immunol. 2006;18:670-5
39. Baldwin DR, Wise R, Andrews JM. et al. Azithromycin concentrations at the sites of pulmonary infection. Eur Respir J. 1990;3:886-90
Author contact
![]() Correspondence to: Yoko Shibata, Department of Cardiology, Pulmonology, and Nephrology, Yamagata University School of Medicine, 2-2-2 Iida-Nishi Yamagata 990-9585, Japan. Tel: +81-23-628-5302; Fax: +81-23-628-5305; E-mail: shibataid.yamagata-u.ac.jp
Correspondence to: Yoko Shibata, Department of Cardiology, Pulmonology, and Nephrology, Yamagata University School of Medicine, 2-2-2 Iida-Nishi Yamagata 990-9585, Japan. Tel: +81-23-628-5302; Fax: +81-23-628-5305; E-mail: shibataid.yamagata-u.ac.jp
Received 2009-7-27
Accepted 2009-10-19
Published 2009-10-23